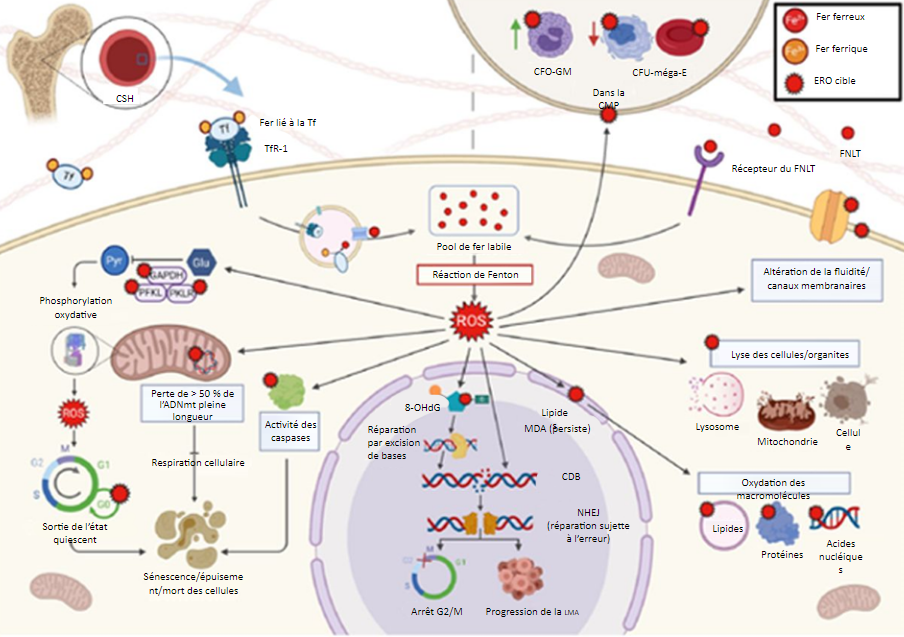
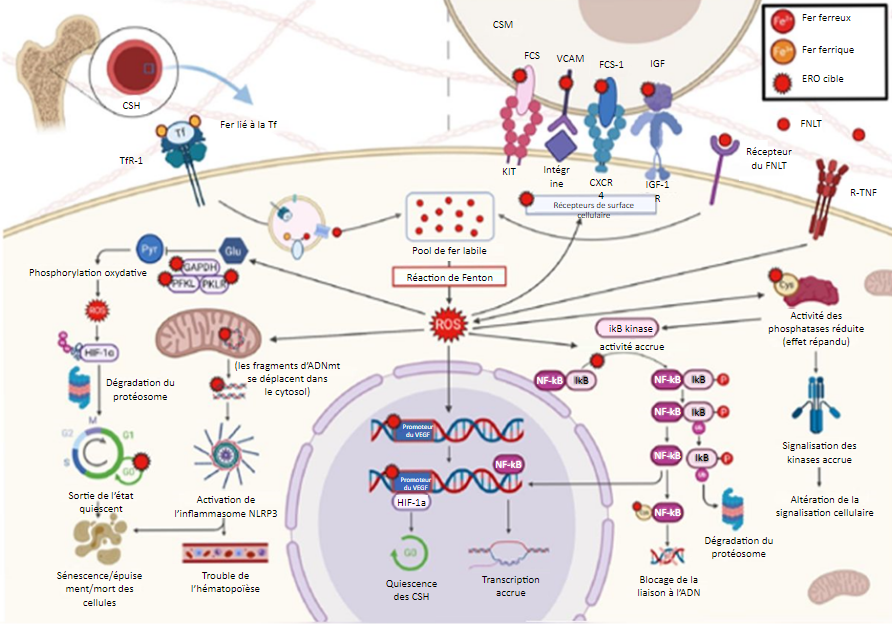
Effets des ERO sur les CSH et leur microenvironnement
a. Dans des conditions normales, le fer pénètre les CSH via le TfR-1. En cas de surcharge de fer, l’entrée non régulée
du FNLT via un récepteur non spécifié conduit à la production d’ERO. Les ERO ont pour cible les enzymes glycolytiques,
ce qui entraîne un passage à la respiration aérobie, et donc la sortie des CSH de l’état quiescent et la promotion du
cycle cellulaire, conduisant ainsi à la sénescence cellulaire, à l’épuisement et/ou à la mort cellulaire. Les ERO endommagent
l’ADNmt, notamment les gènes de la chaîne de transport d’électrons, ce qui empêche la respiration. L’activation des caspases
induite par les ERO mène à l’apoptose. Les ERO engendrent l’oxydation de l’ADN nucléaire, ce qui produit de la 8-OH-dG.
La réparation des dommages oxydatifs au moyen de la réparation par excision de bases endommage davantage l’ADN, ce qui
entraîne des mutations et des CDB. La voie de réparation NHEJ est sujette à l’erreur et d’autres mutations entraînent
un arrêt dans la phase G2/M ou une progression clonale. L’oxydation des lipides produit du MDA, qui persiste et oxyde à son
tour d’autres macromolécules. Les ERO modifient la niche hématopoïétique. Dans la CMP, les ERO déplacent la production vers
les UFC-GM et l’éloignent des UFC-méga-E, ce qui peut exacerber l’anémie du patient.
b. Les ERO modulent l’expression de HIF-1α, un régulateur de l’homéostasie de l’oxygène et un activateur
transcriptionnel, en augmentant son ubiquitination et sa dégradation protéasomique. Un taux élevé de HIF-1α
permet aux CSH de rester en état de quiescence; lorsque le taux de HIF-1α diminue, les CSH sortent de leur
état de quiescence. Les fragments d’ADNmt oxydés activent l’inflammasome NLRP3, qui entraîne ce qui caractérise les
SMD, c’est-à-dire la macrocytose, la prolifération, l’hématopoïèse inefficace et la pyroptose. Les ERO oxydent le
promoteur nucléaire du VEGF, qui recrute et lie les HIF-1α, entraînant ainsi la quiescence des CSH. Ces effets opposés
des HIF-1α peuvent être dus à des taux, à des espèces ou à des emplacements subcellulaires variables des ERO.
Les ERO produits par l’activation du R-TNF inhibent les phosphatases, entraînant une augmentation généralisée de
l’activité des kinases, ce qui modifie la signalisation cellulaire. Par exemple, l’augmentation de l’activité de la
kinase IκB accroît la phosphorylation de l’IκB, un inhibiteur de NF-κB, ce qui entraîne l’ubiquitination et la dégradation
protéasomique de l’IκB, avec pour résultat un NF-κB – un facteur de transcription – libre. Le résidu cystéine de NF-κB,
sensible à l’état redox, subit une glutathionylation oxydative, et la capacité de liaison de NF-κB à l’ADN s’en trouve réduite.
Dans le microenvironnement de la MO, l’expression de protéines de surface spécifiques des CSM a été réduite (FCS, FCS-1,
VCAM-1 et IGF-1, qui interagissent, respectivement, avec KIT, CXCR4, l’intégrine et l’IGF-1R sur les CSH), diminuant ainsi la
liaison des CSM aux CSH et leur soutien.
8-OH-dG, 8-hydroxy-2′-désoxyguanosine; ADN, acide désoxyribonucléique; ADNmt, ADN mitochondrial; CDB, cassures double brin;
CSH, cellules souches hématopoïétiques; CSM, cellules souches mésenchymateuses; CXCR4, récepteur de chimiokine C-X-C de type 4;
E, érythroïde; ERO, espèces réactives de l’oxygène; FCS, facteur des cellules souches; FNLT, fer non lié à la transferrine; G2/M,
transition du cycle cellulaire de la phase G2 à la mitose; GM, granulocyto-monocytaire; HIF-1α, facteur induit par l’hypoxie
1 alpha; IGF-1, facteur de croissance 1 analogue à l’insuline; IGF-1R, récepteur de l’IGF-1; IκB, inhibiteur de NF-κB; KIT,
proto-oncogène KIT, récepteur à activité tyrosine kinase; LMA, leucémie myéloïde aiguë; MDA, malonaldéhyde; méga, mégacaryocyte;
MO, moelle osseuse; NF-κB, facteur nucléaire kappa b; NHEJ, ligature d’extrémités non homologues; NLRP3, protéines composées d’un
domaine de liaison aux nucléotides et d’un domaine riche en leucine de répétition; SMD, syndrome myélodysplasique; TNF, facteur de
nécrose tumorale; UFC, unité formatrice de colonies; VEGF, facteur de croissance de l’endothélium vasculaire.
Kim CH & Leitch HA. Crit Rev Oncol Hematol. 2021;163, avec permission.