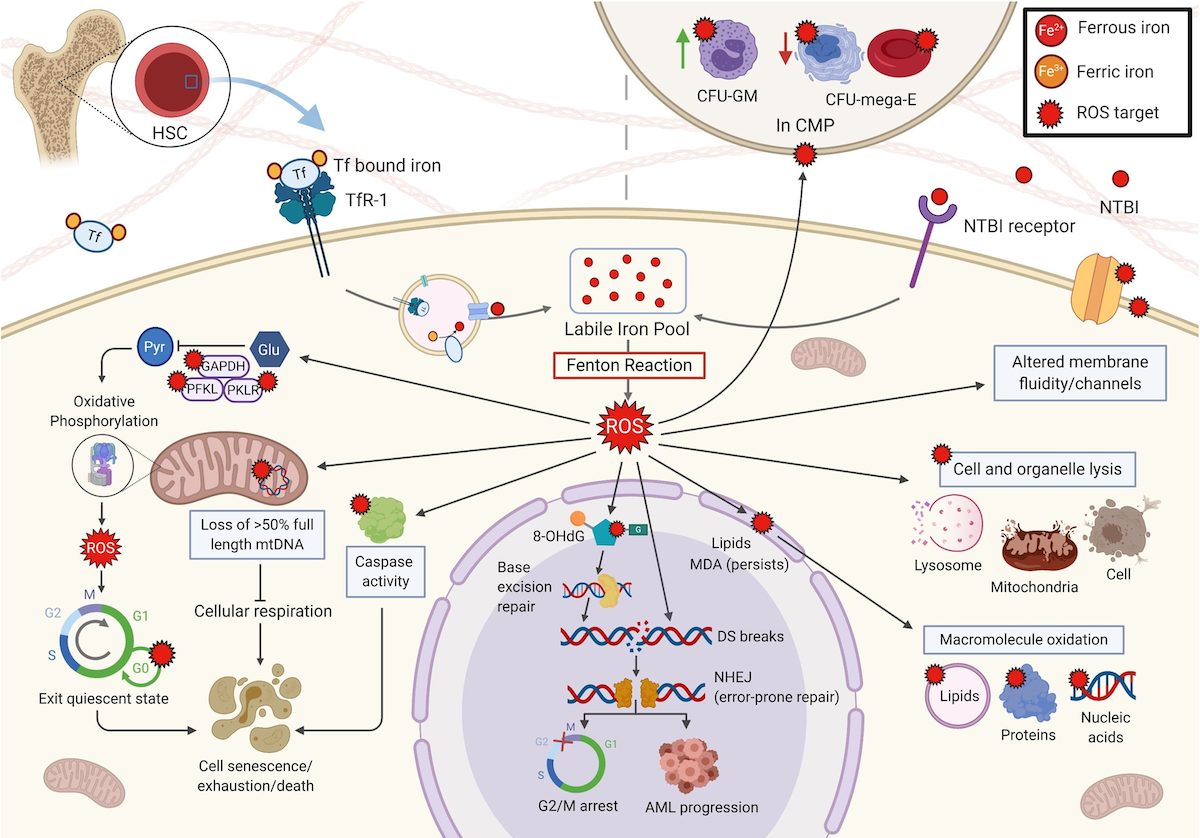
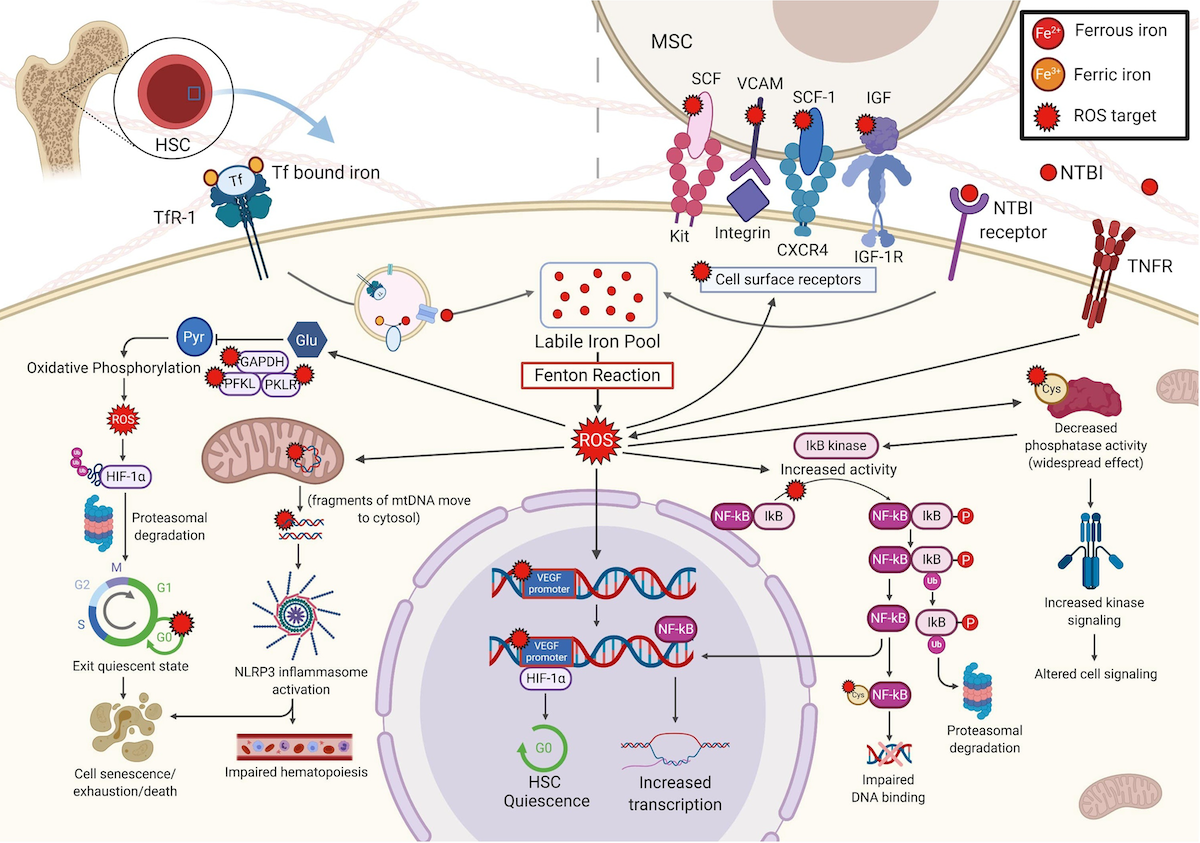
Effects of ROS in the HSC and its microenvironment
a. Iron enters the HSC via TfR-1 under normal conditions. In IOL, unregulated entry of NTBI via an unspecified receptor
leads to ROS production. ROS targets include glycolytic enzymes causing a shift to aerobic respiration, resulting in
exit of HSC from the quiescent state and promotion of cell cycling, leading to cell senescence, exhaustion, and/or cell
death. ROS damage mtDNA, including genes of the ETC, inhibiting respiration. ROS induced caspase activation leads to
apoptosis. ROS oxidizes nuclear DNA, producing 8-OH-dG. Repair of oxidative damage by BER further damages DNA leading to
mutations and DSB. The NHEJ repair pathway is error-prone and further mutations cause arrest at G2/M or clonal
progression. Oxidation of lipids produces MDA, which persists and further oxidizes other macromolecules. ROS alters the
hematopoietic niche. In the CMP, ROS shifts production towards CFU-GM and away from CFU-mega-E, potentially exacerbating
patient anemia.
b. ROS modulate expression of HIF-1α, a regulator of oxygen homeostasis and a transcriptional activator, by
increasing its ubiquitination and proteasomal degradation. High HIF-1α allows HSC to remain quiescent; as
HIF-1α decreases, HSC exit the quiescent state. Oxidized mtDNA fragments activate the NLRP3 inflammasome,
resulting in the MDS features macrocytosis, proliferation, ineffective hematopoiesis, and pyroptosis. ROS oxidizes the
nuclear VEGF promoter, which recruits and binds HIF-1α, leading to HSC quiescence. These opposing effects of
HIF-1α may be due to varying levels, species, or subcellular locations of ROS. ROS produced by activation of the
TNFR inhibits phosphatases, leading to a widespread increase in kinase activity, altering cell signalling. For example,
increased IκB kinase activity increases phosphorylation of IκB, an NF-κB inhibitor, causing IκB ubiquitination and
proteasomal degradation, resulting in free NF-κB, a transcription factor. The redox-sensitive NF-κB cysteine residue
undergoes oxidative glutathionylation, and binding ability of NF-κB to DNA is reduced. In the BME, expression of
specific MSC surface proteins were reduced (SCF, SCF-1, VCAM-1, and IGF-1, which interact, respectively, with KIT,
CXCR4, integrin, and the IGF-1R on HSC), thereby decreasing MSC binding to and support of HSC.
8-OH-dG, 8-hydroxy-2' -deoxyguanosine; AML, acute myeloid leukemia; BER, base excision repair; BM, bone marrow; BME,
bone marrow microenvironment; CFU, colony forming unit; CXCR4, C-X-C chemokine receptor type 4; DSB, double strand
breaks; DNA, deoxyribonucleic acid; E, erythroid; G2/M, growth 2/mitosis transition of the cell cycle; GM,
granulocyte-macrophage; HIF-1α, hypoxia inducible factor-1 alpha, HSC, hematopoietic stem cell; IGF1, insulin like
growth factor 1; IGF-1R, IGF1 receptor; IκB, inhibitor of NF-κB; IOL, iron overload; KIT, KIT Proto-Oncogene, Receptor
Tyrosine Kinase; MDA, malonaldehyde; mega, megakaryocyte; MDS, myelodysplastic syndrome; MSC, mesenchymal stromal cell;
mtDNA, mitochondrial DNA; NF-κB, nuclear factor kappa b; NHEJ, nonhomologous end joining; NLRP3, nucleotide-binding
domain and leucine-rich repeat containing proteins; NTBI, non-transferrin bound iron; ROS, reactive oxygen species; SCF,
stem cell factor; TNFR, tumour necrosis factor receptor; VEGF, vascular endothelial growth factor.
Kim CH & Leitch HA. Crit Rev Oncol Hematol. 2021;163, with permission.