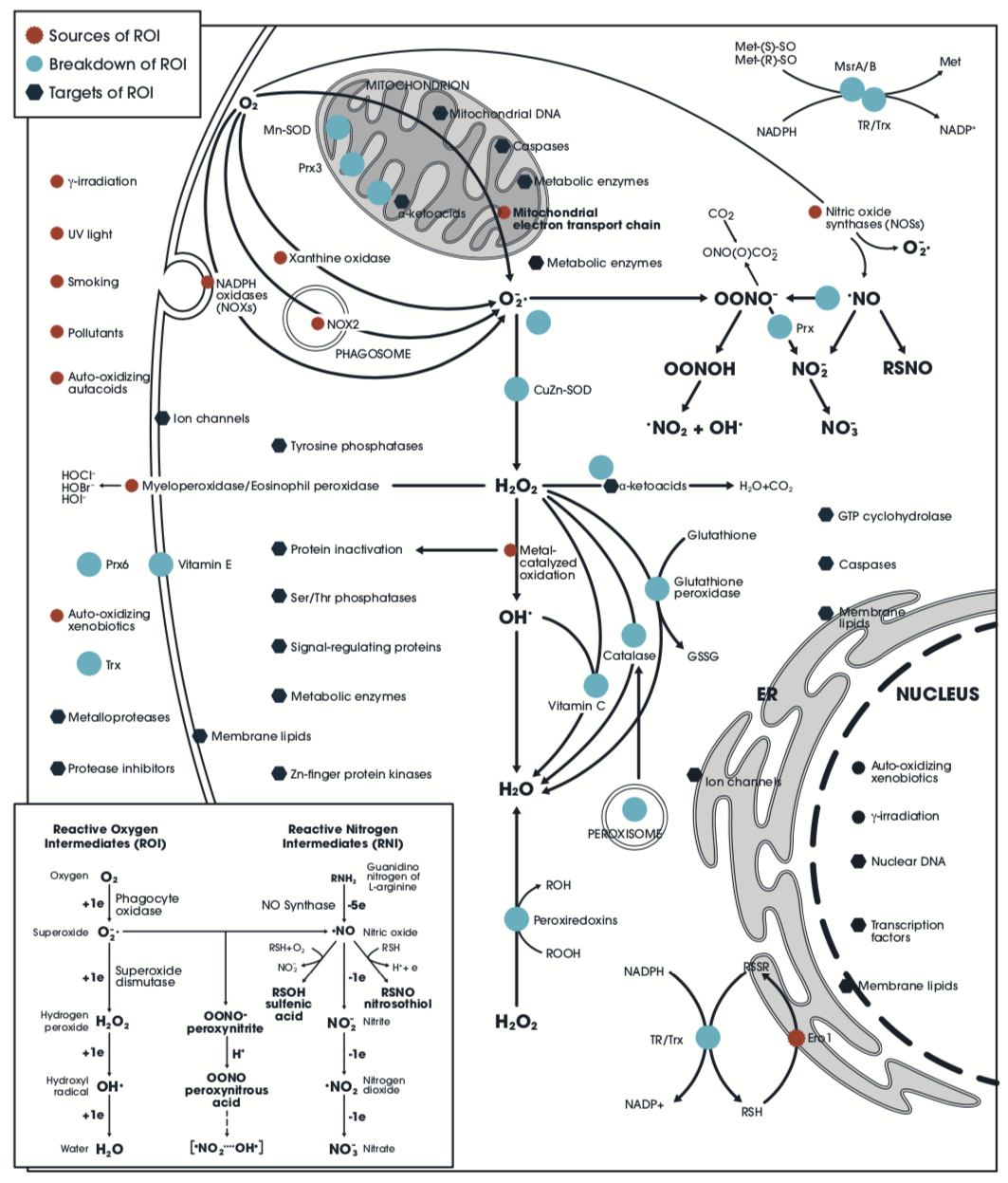
Reactive oxygen intermediates (ROI) and related molecules; their reactions, specificity, sources (red), targets (dark blue), and catabolic routes (light blue; anti-oxidant defenses). For detailed discussion, see Nathan C, & Ding A. Cell. 2010;140(6):951-951.
ROI, ROS, and RNI: ROI are reduction products of O2 en route to H2O production (inset); they include superoxide anion radical (O2−, O2•−), hydrogen peroxide (H2O2), and hydroxyl radical (•OH). Reactive oxygen species (ROS) include ROI, ozone (O2), and singlet oxygen (1O2). ROI and ROS may also include hypochlorous (HOCl) and other acids from halide oxidation. Reactive nitrogen intermediates (RNI) are nitric oxide radical (NO, •NO), nitrogen dioxide radical (•NO2), nitrite (NO2−), and peroxynitrite (ONOO−).
Reactions: Covalent but often reversible, include H2O2 with sulfhydryls e.g. cysteine (RSH) to yield disulfides (RSSR’); and NO with sulfhydryls to yield S-nitrosothiols (RSNO). Reactions with macromolecules involve specific atoms but only some of that type, which is termed atomic specificity. For example, ROI react with sulfur, but usually only in methionine or cysteine such as those in active sites of tyrosine phosphatases. At high levels, ROI are toxic as with any signaling inappropriate in time, place, level, or duration. ROI coordinate signaling pathways through dependence on oxygenation, electron transport, enzyme activity, redox state, and levels of α-ketoacids, NADPH and NADH. Irreversible reactions include hydroxylations, carbonylations, amine halogenations, nitrations, and destruction of iron-sulfur clusters.
Sources: The mitochondrial electron transport chain (ETC) leaks 1%–2% of its electrons as O2−; hypoxia increases ROI by deregulating the ETC. NO, OONO−, and elevated intracellular Ca2+ also promote ROI generation. A major ROI source is NADPH oxidases (NOXs), activated in response to cytokines. Nitric oxide synthases (NOSs) produce O2− when uncoupled by hypoxia or oxidation. Iron-catalyzed oxidations include the Haber-Weiss and Fenton reactions (ferric- and ferrous-catalyzed generation of •OH). Xanthine oxidase is a source of ROI during ischemia-reperfusion. γ-irradiation generates •OH. UV light generates 1O2 and induces NOS2. Air pollutants contain ROS and organic radicals.
Targets: Tyrosine and serine/threonine phosphatases have active-site cysteine residues subject to reversible, oxidative inactivation, affecting kinase pathways (e.g. MAPK). ROI oxidize cysteines that coordinate Zn2+ in Zn2+-finger proteins, leading to activation of protein kinase C. Metalloproteases undergo oxidation of cysteine residues that maintain the inactive state. Caspases are inhibited through cysteine, blocking apoptosis. Cysteine oxidation affects signal-regulation by heat shock proteins. Transcription factors undergo oxidative activation (Nrf2, NF-κB). Metabolic enzymes are inhibited by ROI. A major impact of H2O2 is oxidative decarboxylation of α-ketoacids (pyruvate, α-ketoglutarate, and oxaloacetate of the citric acid cycle). Proteins are inactivated through metal-catalyzed oxidation and carbonylation of arginine, lysine, proline, and threonine. Protease inhibitors that restrain tissue damage during inflammation inactivated by oxidation. ROI activate ion channels, nuclear and mitochondrial DNA are targets for ROI-induced mutagenesis, and ROI actions on lipids has widespread consequences.
Breakdown: CuZn-superoxide dismutase (SOD) in the cytosol and Mn-SOD in mitochondria convert O2− to H2O2. Catalase in peroxisomes acts on H2O2. The glutathione (GSH) cycle is driven by NADPH; this reduces disulfide (S-S) bonds, donates an electron (e-) to peroxiredoxins (Prx’s) or ascorbate, and reverses protein S-nitroso bonds. The thioredoxin (Trx) cycle is similarly driven by NADPH; it reduces S-S bonds and donates e- to Prx’s, and also methionine sulfoxide (MetSO) reductases and ribonucleotide reductase. Prx 1–6 reduce H2O2, organic peroxides, and ONOO−. MetSO reductases detoxify H2O2, as do α-ketoacids. Ascorbate reduces H2O2 and scavenges •OH while tocopherols (vitamin E) react with lipid radicals induced by ROS.